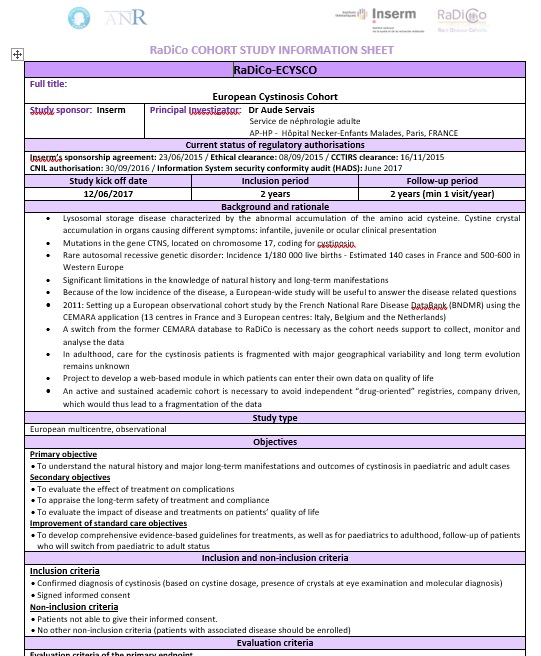
RaDiCo-SED-VASC
Cohorte RaDiCo-SED-VASC
Cohorte nationale sur le syndrome d’Ehlers-Danlos vasculaire
Investigateur Principal : Xavier Jeunemaître
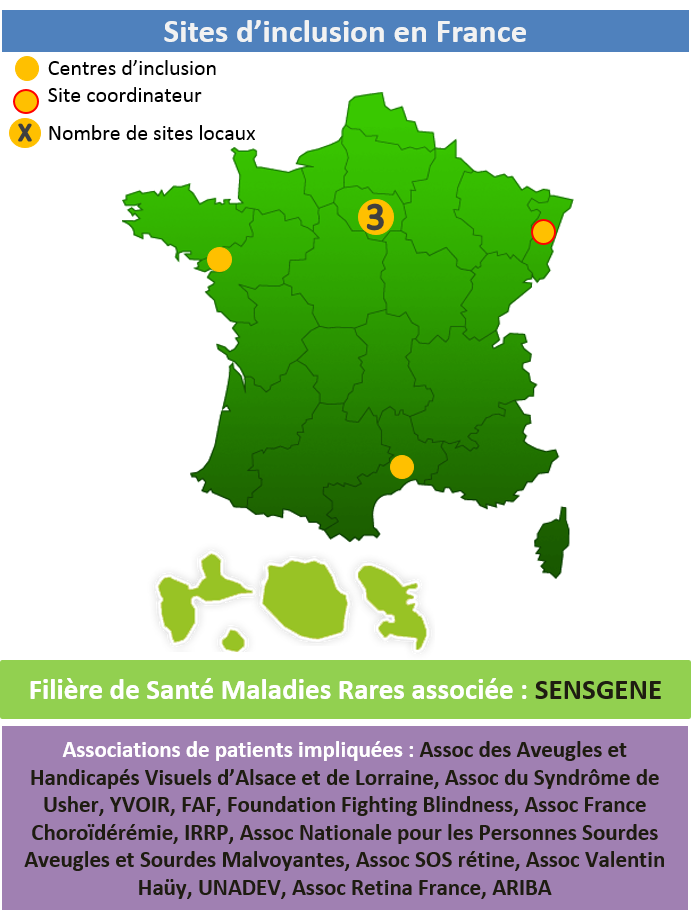
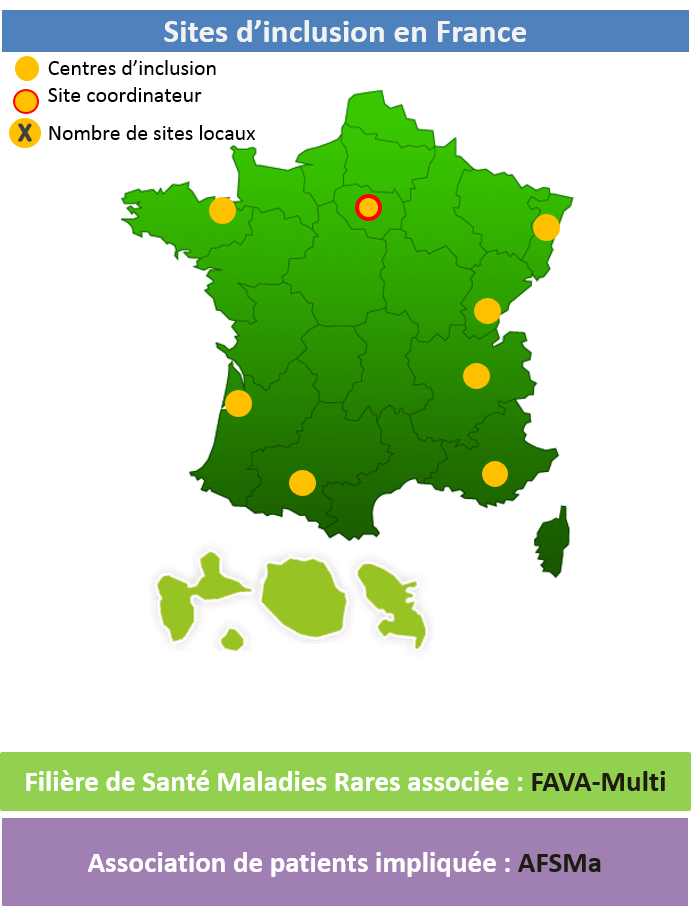
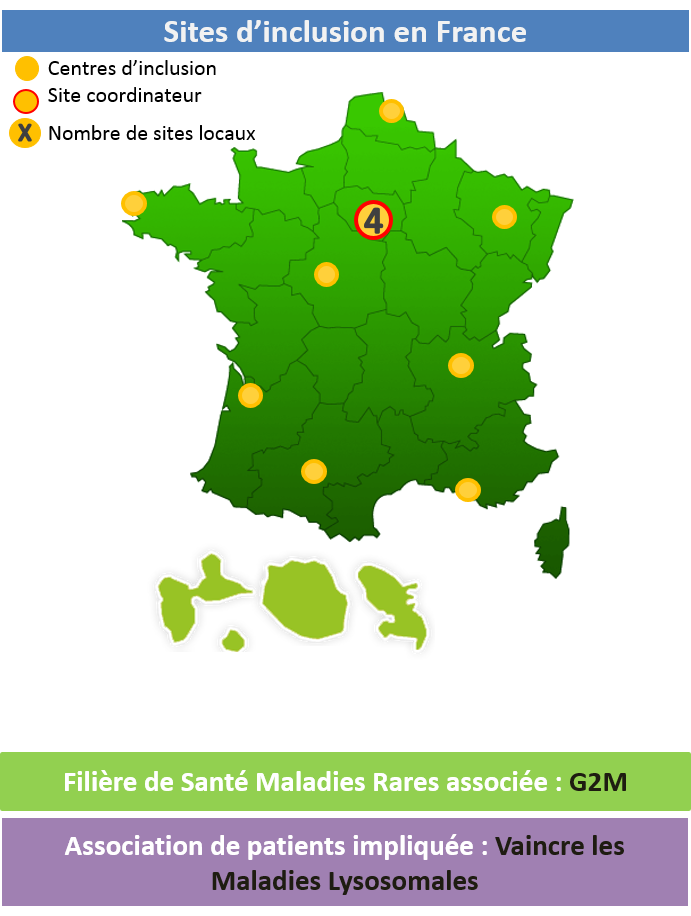
Filière de Santé Maladies Rares associée : FAVA-Multi
Contact Comité Scientifique : Cette adresse e-mail est protégée contre les robots spammeurs. Vous devez activer le JavaScript pour la visualiser.
Description et besoins :
Le syndrome d’Ehlers-Danlos vasculaire (SEDv), est une maladie autosomique dominante rare (prévalence estimée de l’ordre 1/200 000, soit environ 300-400 familles en France) causée par des mutations dans le gène COL3A1 qui code pour le collagène de type III, un composant essentiel de la matrice extra-cellulaire. L’expression de ce collagène défectueux peut se traduire par des lésions artérielles (anévrisme, dissection localisée, rupture) ou digestives (perforation colique) qui sont la source de la majorité des évènements morbides ou fatals de cette pathologie. Les premières complications apparaissent chez le jeune adulte à un âge moyen de 29 ans. Plus de 130 patients SEDv formellement diagnostiqués (mutation du gène COL3A1) sont suivis par le Centre de Référence des Maladies Vasculaires Rares (CRMVR) et près de 135 patients sont pris en charge par les 15 Centres de Compétence affiliés formant ainsi la plus grande cohorte active pour cette pathologie au plan mondial. Ceci a permis la mise en place d’études (i) démontrant le bénéfice du céliprolol dans la prévention des ruptures artérielles, (ii) démontrant l’association de certaines mutations avec la sévérité de la maladie, (iii) d’identification de biomarqueurs associés à la sévérité des lésions artérielles. Une base de données permettant un suivi épidémiologique longitudinal de la pathologie est indispensable.
Bénéfice attendu :
La cohorte SED-VASC a pour objectifs
- d’améliorer la prise en charge des patients par (i) l’étude de l’histoire naturelle précise de la maladie et en particulier les liens entre les différents signes majeurs et mineurs de la pathologie, les différents types de complications (artérielles, digestives, pulmonaires, utérines), l’étude fine et prospective des relations génotype-phénotype, (ii) la validation prospective en ouvert de l’effet bénéfique du traitement de référence (celiprolol), en particulier sur la récidive/cicatrisation des ruptures artérielles localisées et la morbi?mortalité associée,
- d’améliorer le développement de nouvelles options thérapeutiques par (i) la constitution d’un registre national de la pathologie Interfacé avec le minimum data set de BaMaRa (base maladies rares) et ayant satisfait à tous les critères technico-réglementaires et dont les variables soient échangeables à l’international, (ii) la facilitation du recrutement pour des essais thérapeutiques prospectifs nationaux et internationaux,
Engagement de RaDiCo :
La contribution et l’expertise apportées par RaDiCo portent sur la structuration de l’étude de la cohorte SED-VASC avec le développement de bases de données interopérables et d’outils d’information performants et innovants, pour permettre le recueil et la corrélation de données de nature et d’origine variées (clinique, imagerie, qualité de vie du patient, génétique, pharmaco-économique…). De plus, l’expertise de RaDiCo pour étendre à l’international un tel programme est une chance unique pour la mise en place d’une cohorte fédérative européenne.Enfin, l’un des atouts majeurs de RaDiCo réside dans le développement de standards de qualité homogènes pour le recueil, la mesure et l’analyse des données dans ce domaine des maladies rares.